 Due to Customs restrictions, we only accept orders from educational institutions within the Continental United States, Alaska or Hawaii.
Due to Customs restrictions, we only accept orders from educational institutions within the Continental United States, Alaska or Hawaii. What Is Sickle Cell Anemia?
What Is Sickle Cell Anemia?
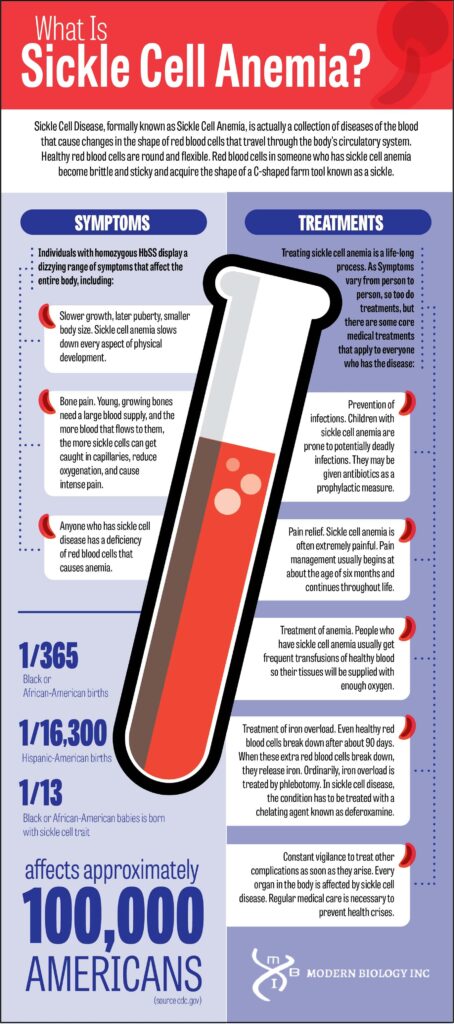
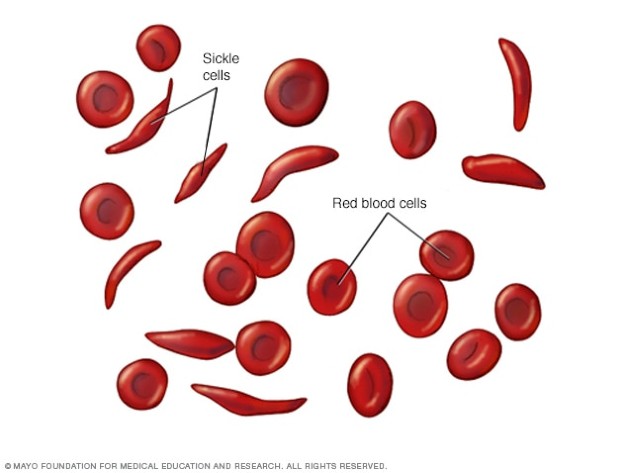
Sickle Cell Disease, formally known as Sickle Cell Anemia, is actually a collection of diseases of the blood that cause changes in the shape of red blood cells that travel through the body’s circulatory system. Healthy red blood cells are round and flexible. Red blood cells in someone who has this condition become brittle and sticky and acquire the shape of a C-shaped farm tool known as a sickle.
Healthy red blood cells have a 90-day life span, while sickle cells have a much shorter life span. As a result, the body has a constant shortage of red blood cells, causing anemia, or shortage of oxygen to body tissues. In addition to anemia, these cells get stuck and form clots in smaller blood vessels, resulting in chronic, often severe pain, along with increased risk of heart attack, pulmonary embolism, deep vein thrombosis, and stroke.
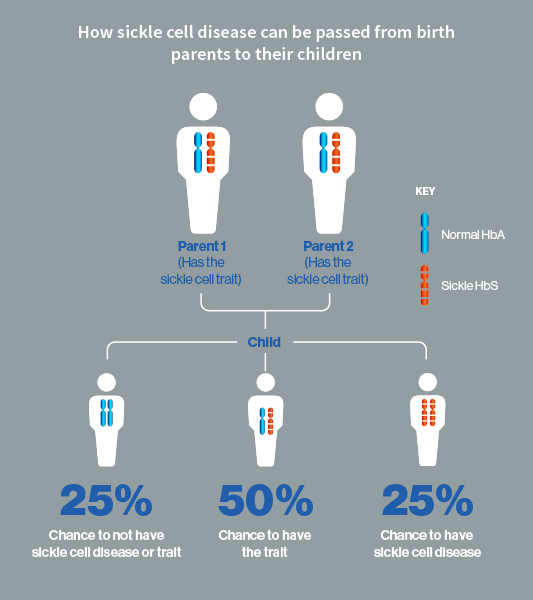
Homozygous HbS disease is an autosomal recessive hereditary condition most common among people of African or Mediterranean descent. The most common form of this condition found in the United States is homozygous HbS disease (HbSS).
The reason this gene has persisted among people of African and Mediterranean heritage is that having just one of two possible genes for this condition confers resistance to Malaria. Compound heterozygotes for this disease were seen as healthier than other individuals who did not have any of these genes at all and had more children, however this does not mean that people who carry just one of these genes are perfectly healthy. In modern times, there is no competitive advantage for most Americans to be resistant to malaria, so when both parents carry at least one gene for this condition, their children are unfortunately at risk for developing the full-blown form of the disease.
Symptoms of Sickle Trait Condition
The predominant difference between Sickle Trait Condition and Sickle Cell Disease comes from the number of recessive traits received from the individual’s parents. If a person is born with a single recessive trait, they have Sickle Trait Condition, whereas an individual born with two recessive traits will have the full disease.
People who have just one of these genes exhibit varying degrees of the condition. These individuals do not necessarily develop anemia normally, but because about 40% of their red blood cells show sickle cell morphology, they don’t handle oxygen stress like high altitude, lung disease, or exercise very well and become hypoxic more easily than healthy people. Additional symptoms include watery urine and blood in the urine because of the effects of these cells in the fine blood vessels of the kidneys.
Receiving both of the HbSS genes results in the disease, also known as anemia. Affected individuals display a wide range of symptoms associated with their condition.
Symptoms of Sickle Cell Disease
Individuals with homozygous HbSS display a dizzying range of symptoms that affect the entire body, including:
Slower growth, later puberty, smaller body size. This condition slows down every aspect of physical development.
Bone pain. Young, growing bones need a large blood supply, and the more blood that flows to them, the more sickle cells can get caught in capillaries, reduce oxygenation, and cause intense pain. Children who have this disease are subject to frequent rounds of bone pain as their bones continue to grow.
Anemia. Anyone who has sickle cell disease has a deficiency of red blood cells that causes anemia. The resulting poor oxygenation of the bloodstream makes heavy exercise, such as running, playing, and participating in sports, difficult or impossible in teenagers and adults who have the disease. Sometimes children are able to compensate for hypoxia well enough to spend some time on the playground.
People who have anemia are prone to health crises throughout their lives. Children with the disease may develop splenic sequestration in which the spleen becomes engorged with blood, so anemia becomes even worse. This condition is potentially life-threatening. Infections of the bloodstream pose a danger to children, and adults are at risk for stroke, pulmonary hypertension, and, in men, a condition known as priapism.
Over the last 30 years, infants born in the United States have been tested for a variety of different diseases including Sickle Cell Disease. It is important for an early diagnosis to help prevent affected children from contracting other dangerous diseases that commonly occur during childhood. However, it is still possible to be a carrier of the disease and subject to the combination of symptoms known as Sickle Trait Condition.
What are the Treatments for Sickle Cell Anemia?
Treating this condition is a life-long process. As Symptoms vary from person to person, so too do treatments, but there are some core medical treatments that apply to everyone who has the disease:
Prevention of infections. Children with this condition are prone to potentially deadly infections. They may be given antibiotics as a prophylactic measure.
Pain relief. Sickle cell anemia is often extremely painful. Pain management usually begins at about the age of six months and continues throughout life.
Treatment of anemia. People who have this condition usually get frequent transfusions of healthy blood so their tissues will be supplied with enough oxygen. Frequent transfusions, however, lead to another problem.
Treatment of iron overload. Even healthy red blood cells break down after about 90 days. When these extra red blood cells break down, they release iron. Iron levels can become so high they cause a disease known as acquired hemochromatosis, or iron overload. The extra iron can “rust” tissues in the liver, pancreas, skin, and brain. Ordinarily, iron overload is treated by phlebotomy, drawing blood to reduce the body’s iron load. In this disease, the condition has to be treated with a chelating agent known as deferoxamine, which can cost up to $20,000 per month.
Constant vigilance to treat other complications as soon as they arise. Every organ in the body is affected by this disease. Regular medical care is necessary to prevent health crises.
So far, the use of genetic engineering has cured at least one person of Sickle Cell Disease. A gene that creates healthy hemoglobin is inserted into the patient’s stem cells, and in time these cells take over the production of red blood cells and symptoms go away. The condition can also be treated with allogeneic marrow transplantation (replacement of stem cells in the bone marrow with stem cells from another person). This procedure is successful about 84% of the time.
Get Started Studying Sickle Cell Anemia
Modern Biology offers an exciting experiment geared for the teaching laboratory to learn more about this condition and use the actual clinical method to test for the disease using real hemoglobin samples. The experiment also features an optional opportunity for students to test their own blood as well.
Genetic Engineering programs are also available through Modern Biology. These multi-lab programs give students a first-hand look into how many diseases, including Sickle Cell Disease, can be treated. Visit Modern Biology for complete courses and all your classroom biology supplies.